Основные способы классификации микроорганизмов и филогения, основанная на изучении последовательностей рРНК

Следует подчеркнуть, что в других классификационных схемах, принятых, например, в зоологии или ботанике, критерии отнесения к роду или виду могут существенно отличаться по этому признаку. Далее определение видового названия ведется традиционными микробиологическими методами. Для уточнения таксономической принадлежности проводят ДНКДНК гибридизацию, которая дает > 30% совпадения в пределах рода и… Читать ещё >
Основные способы классификации микроорганизмов и филогения, основанная на изучении последовательностей рРНК (реферат, курсовая, диплом, контрольная)
В настоящее время существует несколько основных способов классификации живых объектов, в том числе микроорганизмов.
Формальная нумерическая классификация считает все признаки организма одинаковыми по значимости. Учитываемые критерии должны быть альтернативными, т. е. присутствовать (+) или отсутствовать (-) у конкретного объекта. Точность помещения его в данную группу будет зависеть от полноты изучения организма. Для количественной оценки степени сходства и различия объектов разработаны специальные компьютерные программы, сравнивающие организмы по набору исследованных признаков. Сходные организмы объединяются в кластеры.
Для морфофизиологической классификации необходимо изучить не только совокупность морфологических признаков, но и особенности метаболизма организмов. При этом учитывают разную значимость применяемых критериев: некоторые свойства считают обязательными, значимыми для объекта, а другие могут присутствовать в разной степени или совсем отсутствовать. Результаты всех необходимых тестов применяют при работе с определителем. В настоящее время для идентификации прокариотических микроорганизмов исследователи пользуются определителем, носящим имя американского бактериолога Берджи, предложившего в 1923 г. основы такой классификации.
Молекулярно-генетическая классификация предполагает анализ строения молекул важных биополимеров. Такая молекула должна быть консервативной и значимой для основополагающего жизненного процесса. Профессор Иллинойского университета Карл Вез предложил взять за основу рибосомальную РНК (16S рРНК для прокариот, 18S рРНК для эукариотических организмов). Эта молекула входит в состав рибосом, которые у всех живых существ отвечают за важнейший жизненный процесс — синтез белка. Аппарат синтеза белка незначительно меняется во времени, так как любое сколько-нибудь существенное нарушение может привести к гибели клетки. Поэтому в молекулах рРНК разных организмов большинство нуклеотидов неизменно, а изменяющаяся в процессе эволюции часть уникальна для конкретного организма. 16S рРНК состоит из 1500 нуклеотидов, из которых 900 — консервативны, т. е. она обладает достаточно большой, но не чрезмерной информацией и может считаться своеобразным биологическим генетическим «хронометром». Сравнивая с помощью специальных компьютерных программ нуклеотидные последовательности этой молекулы у разных организмов, можно получить группы сходства биологических объектов, отражающие их родственные связи и эволюционное развитие. На основе множества сравнений было построено филогенетическое древо, где все представители живого мира разделены на три больших домена (империи, надцарства): Bacteria, Archaea и Eukarya (рис. 10.1). Домены Bacteria и Archaea содержат только прокариотические организмы, а домен Eukarya объединяет всех эукариот — как одноклеточных, так и многоклеточных, включая человека.
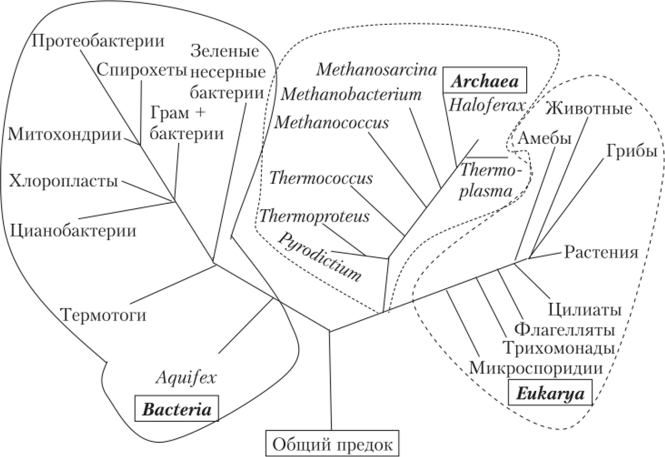
Рис. 10.1. Отдельные филогенетические группы филогенетического древа, основанного на сравнении 16 (18)S рРНК.
При анализе рибосомальных РНК из митохондрий и хлоропластов было показано, что они имеют прокариотное симбиотическое происхождение. Нуклеотидные последовательности изученных организмов исследователи направляют во всемирный компьютерный гепбанк (GenBank), данные которого предназначены для проведения сравнения с последовательностями каждого вновь выделенного организма.
В настоящее время для идентификации конкретного микроорганизма выделяют его чистую культуру и сначала проводят анализ нуклеотидной последовательности 16S рРНК. Сравнение с уже известными последовательностями позволяет поместить микроорганизм в один из кластеров на филогенетическом древе, учитывая следующие критерии сходства последовательностей нуклеотидов: «вид» > 97%, «род» > 94%, «семейство» > 92%, «порядок» > 90%, «класс» > 85%, «филум» > 80%. Данные критерии названы операционными таксономическими единицами (OTU). Значения процентов, их определяющие, являются «договорными», общепринятыми в международном научном сообществе для выделенных в настоящее время культивируемых прокариот и клонов некультивируемых бактерий и архей.
Следует подчеркнуть, что в других классификационных схемах, принятых, например, в зоологии или ботанике, критерии отнесения к роду или виду могут существенно отличаться по этому признаку. Далее определение видового названия ведется традиционными микробиологическими методами. Для уточнения таксономической принадлежности проводят ДНКДНК гибридизацию, которая дает > 30% совпадения в пределах рода и > 70% — в пределах вида. Нуклеотидная последовательность вновь выделенного микроорганизма должна быть направлена в генбанк для пополнения базы данных.
Для выявления родственных связей микроорганизмов более узких групп иногда используют и другие молекулы. Например, для дышащих организмов применяют определение последовательности аминокислот в белке цитохрома с. У ряда прокариотических и эукариотических микроорганизмов определена полная последовательность ДНК в хромосомах.
Для более четкой дифференцировки микроорганизмов на уровне рода и вида предложено применять полифилетическую (полифазную) таксономию, когда наряду с определением последовательностей нуклеотидов используют информацию разных уровней, вплоть до экологического. При этом проводят предварительный поиск групп схожих штаммов и определение филогенетических позиций этих групп, фиксируют различия между группами и их ближайшими соседями и собирают данные, позволяющие дифференцировать группы.