Фенилкетонурия (ФКУ). Наследственные болезни обмена веществ

У детей с фенилкетонурией наблюдается повышенный уровень в моче метаболитов ФА. Увеличение в физиологических жидкостях содержания ФА и недоокисленных продуктов его метаболизма приводит к поражению нервной системы. Определенная роль в этих нарушениях принадлежит дисбалансу аминокислот (дефицит тирозина, который в норме активно участвует в построении белкового компонента миелина). Демиелинизация… Читать ещё >
Фенилкетонурия (ФКУ). Наследственные болезни обмена веществ (реферат, курсовая, диплом, контрольная)
Впервые описана в 1934 г. Folling под названием «фенилпировиноградная имбецильность». Тип наследованияаутосомно-рецессивный. Частота заболевания составляет 1: 10 000- 1: 20 000 новорожденных. Пренатальный диагноз возможен при использовании генетических зондов и биопсии ворсин хориона.
К развитию классической клинической картины при ФКУ приводит недостаточность фенилаланингидроксилазы и недостаточность редуктазы дигидроптерина- 2-го фермента, обеспечивающего гидроксилирование фенилаланина. Их недостаток приводит к накоплению фенилаланина (ФА) в жидких средах организма (схема 1). Как известно, ФА относится к незаменимым аминокислотам. Поступающий с продуктами питания и не используемый для синтеза белка, он распадается по тирозиновому пути. При ФКУ наблюдается ограничение превращения ФА в тирозин и, соответственно, ускорение его превращения в фенилпировиноградную кислоту и другие кетоновые кислоты.
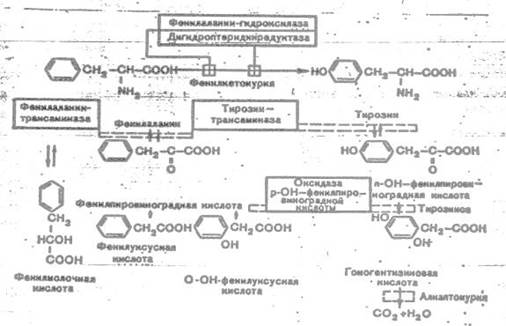
Схема 1. Варианты нарушений метаболизма фенилаланина.
Существование различных клинико-биохимических вариантов ФКУ объясняется тем, что фенилаланингидроксилаза является частью мультиферментной системы.
Различают следующие формы ФКУ:
- 1. Классическая
- 2. Скрытая.
- 3. Атипичная.
Развитие атипичных и скрытых форм ФКУ связывают с недостаточностью фенилаланинтрансаминазы, тирозинтрансаминазы и оксидазы парагидроксифенилпировиноградной кислоты. Атипичная ФКУ обычно не сопровождается поражением нервной системы в результате позднего развития ферментативного дефекта.
У женщин с фенилкетонурией возможно рождение детей с микроцефалией, задержкой умственного развития, нарушениями развития мочевыделительной системы, поэтому необходимо назначение дието;
терапии во время беременности.
Клинические симптомы у больных ФКУ При рождении ребенок с фенилкетонурией выглядит здоровым. Заболевание у этих детей проявляется на первом году жизни.
- 1. Интеллектуальный дефект. Нелеченный ребенок теряет около 50 баллов IQ к концу 1-го года жизни. У больных не выявляется зависимости между уровнем ФА и степенью интеллектуального дефекта.
- 2. Судорожный синдром (4 50%), экзема, гипопигментация.
- 3. Нарушение координации движения.
- 4. Задержка развития статических и двигательных функций.
- 5. Поражение пирамидных путей и стриопаллидарной системы. Клинические проявления классической ФКУ редко встречаются в странах, в которых действует программа неонатального скрининга на это заболевание.
У детей с фенилкетонурией наблюдается повышенный уровень в моче метаболитов ФА. Увеличение в физиологических жидкостях содержания ФА и недоокисленных продуктов его метаболизма приводит к поражению нервной системы. Определенная роль в этих нарушениях принадлежит дисбалансу аминокислот (дефицит тирозина, который в норме активно участвует в построении белкового компонента миелина). Демиелинизация является характерным патоморфологическим признаком фенилкетонурии. Нарушение соотношения аминокислот в крови приводит к нарушению уровня свободных аминокислот в головном мозге, что вызывает слабоумие, гиперкинезы и другие неврологические симптомы.
Пирамидные симптомы обусловлены нарушением процессов миелинизации. Избирательный характер поражения нервной системы объясняется особенностями миелинизациипоражаются наиболее молодые в филогенетическом отношении отделы, выполняющие сложные и дифференцированные функции. С недостаточным образованием меланина из тирозина связывают голубой цвет глаз, светлую кожу. Запах «плесени» («мышиный», «волчий») объясняется наличием фенилуксусной кислоты в моче. Кожные проявления (экссудативный диатез, экземы) связаны с выделением аномальных метаболитов. Недостаточность образования адренергических гормонов из тирозина приводит к артериальной гипотонии.
Необходимо отметить, что при ФКУ в патологический процесс вовлекается печень, но характер морфологических расстройств не является специфичным: выявляются признаки тканевой гипоксии, нарушения окислительной и белоксинтезирующей функции, перегрузка липидами. Наряду с этим наблюдаются компенсаторно-приспособительные изменения: высокое содержание гликогена, гиперплазия митохондрий. Генерализованную гипераминоацидемию при ФКУ можно объяснить вторичным нарушением метаболизма аминокислот в связи с повреждением гепатоцитов, т. к. многие ферменты, участвующие в аминокислотном обмене, локализуются в печени.
У нелеченых больных с классической ФКУ наблюдается значительное снижение концентрации катехоламинов, серотонина и их производных в моче, крови, ликворе. Поэтому в комплексном лечении ФКУ необходима промедиаторная коррекция, так как парциальный интеллектуальный дефект может быть связан с нейромедиаторными нарушениями.
Критерии диагностики классической формы фенилкетонурии:
- 1. Уровень ФА в плазме выше 240 ммоль/л.
- 2. Вторичный дефицит тирозина.
- 3. Повышенный уровень в моче метаболитов ФА.
- 4. Сниженная толерантность к полученному внутрь ФА.